
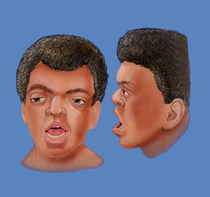
Fig.1- Aparência geral de uma pessoa afectada com Síndrome de Apert
Descrição
Em 1906 Apert publicou um relato de nove casos sobre a doença conhecida como Síndrome de Apert. Trata-se de uma doença de carácter autossómico dominante- o que significa que se um dos progenitores tiver a doença o risco de ocorrência será de 50%.No entanto, já foram relatados casos em que nenhum progenitor apresentava Síndrome de Apert. Uma mutação no gene do receptor 2 do factor de crescimento do fibroblasto (célula do tecido conjuntivo formadora dos tecidos fibrosos do organismo) , mapeado no cromossoma 10q25-10q26, é a responsável pela doença. A incidência da síndrome é de aproximadamente 1 em cada 65.000 nascimentos. Quanto maior a idade do pai maior a probabilidade de o filho ser portador da doença. De referir que esta é uma doença que ocorre no período da fecundação e que factores de causas ambientais (como por exemplo a ingestão de bebidas alcoólicas por parte da mãe durante a gravidez) não interferem no desenvolvimento da mesma.
Características
Os portadores de Síndrome de Apert têm uma aparência típica, podendo apresentar: face achatada, má formação específica do crânio(craniossinostose-fechamento prematuro das suturas ósseas da cabeça, impedindo o crescimento normal da cabeça),má formação de mãos e pés (sindactilia-união de um ou mais dedos formando uma membrana única), nariz estreito e pequeno, orelhas com implantação baixa, fissura palatina, gengivas espessadas, má formação dentária para além de outras diversas alterações funcionais que variam de indivíduo para indivíduo. A maioria dos portadores apresenta deficiência mental com atraso no desenvolvimento.

Fig.2- Criança portadora da síndroma, com sindactilia visível nas mãos
Diagnóstico
O diagnóstico da Síndrome de Apert é clínico, não havendo necessidade de estudos laboratoriais para comprovação da doença. As anomalias craniofaciais podem ser avaliadas por uma tomografia computorizada, ajudando assim no diagnóstico.
{kind=link}
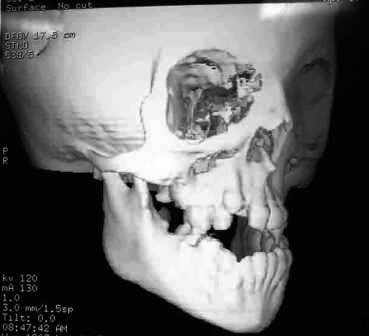
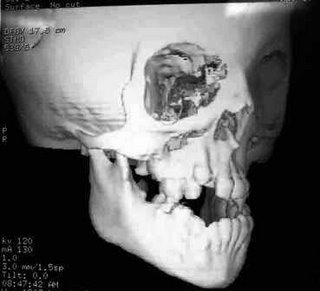 Fig.3-Tumografia evidenciando deformações no crânio do paciente e mal formação dentária.
Fig.3-Tumografia evidenciando deformações no crânio do paciente e mal formação dentária.Tratamentos
O tratamento está dividido em duas partes: tratamento relacionado com a deficiência mental e tratamento de outras características por meio cirúrgico.
Tratamento relacionado com a deficiência mental
Esta doença exige cuidados precoces e enérgicos. Crianças portadoras da síndrome necessitam de ser estimuladas logo após o diagnóstico, pois as deficiências no desenvolvimento básico podem ser remediadas. A Educação Física adaptada tem vindo a ser valorizada como uma das condições essenciais para o desenvolvimento motor, intelectual, social e efectivo dos doentes.

Tratamento por via cirúrgica
Correcção de sindactilia- através de uma cirurgia é possível separar os dedos, cortando para isso a membrana que os liga. A imagem seguinte mostra uma mão com dois dedos juntos e uma outra já depois da operação, onde é visível o corte da membrana que unia os dedos.

Correcção da craniossinostose- intervenção cirúrgica que tem como objectivo abrir as suturas cranianas para um desenvolvimento normal do crânio do portador. Na imagem seguinte é possível observar a diferença de um doente que sofreu este tipo de operação. A primeira figura é antes da operação; a segunda é depois.

Correcções dentárias e á fissura palatina são outras cirurgias que contribuem para o tratamento da Síndrome de Apert.
Nenhum comentário:
Postar um comentário